


Robert K. Koenekoop, MD, MSc, PhD, FRCS(C), FARVO
Montreal Children’s Hospital, Chief Paediatric Ophthalmology at the McGill University Health Center (MUHC)
Professor of Paediatric Surgery, Human Genetics and Ophthalmology at McGill University
Director of the laboratory for Retinal Genetics and Therapeutics Director of the laboratory for Retinal Genetics and Therapeutics
1001 Blvd Decarie, Montreal, Québec, Canada H4A 3J1
Emails: robkoenekoop@hotmail.com, For IRD patients email appointments: fightingblindness@cmbmed.com
Biographie/Biography
RESEARCH PROFILE: Dr. Robert Koenekoop (Rob) was born in Stockholm, Sweden but went to High school and the University of Utrecht in The Netherlands. After Bachelor’s and Master’s degrees in Biology, Vegetation Science and Population Biology, a Fulbright fellowship from Amsterdam brought him to the USA for a PhD in Molecular Biology. His wife then brought him to Quebec, Canada and Medical school studies at the University of Toronto and McGill University followed. He saw the light in the retina clinic and finished his residency in Ophthalmology at McGill and his Ocular Genetics and Paediatric Ophthalmology Fellowships at Johns Hopkins University. For the past 25 years he has devoted his research career to discovering new retinal genes for childhood blindness due to retinal degenerations, and more recently in testing the safety and efficacy of new therapies for these same diseases, with some very important early successes. He has a broad background in human clinical trials and drug development, molecular genetics, clinical and paediatric ophthalmology, retinal degenerations, childhood blindness
research and data analyses. In the past few years, in international collaborations, he has been able to discover 15+ new genes for childhood blindness due to retinal degenerations. This work was supported by grants from NIH (NEI), CIHR, Fighting Blindness Canada, The MCH foundation, Telethon of stars, the FRSQ and Reseau Vision. This led to the publication of 150+ peer-reviewed papers. He is now the principal investigator (PI) at McGill, the Montreal Children’s Hospital and the MUHC Center for Innovative Medicine (CIM) for human clinical trials to test new drugs, new genes, gene editing, new genetic methods and other modalities to combat blindness due to photoreceptor diseases. In his free time, he bikes 365 days per year and keeps a healthy diet.
CLINICAL PROFILE : In Canada, Dr. Koenekoop sees Paediatric and Inherited Retinal Degeneration patients 5 days per week, at the new Glen Eye clinic (MUHC), at the new Children’s Clinic and at the new Mohawk Eye clinic. IRD patients in these clinics receive extensive workups, deep genetic testing and genetic and treatment counselling.
***
Résumé/Abstract
Titre:
Inherited Retinal Degenerations (IRDs) are devastating retinal photoreceptor degenerations, leading to complete blindness. Almost 6 million people around the world have IRDs and carry mutations in one of over 300 retinal genes. Twenty genes are commonly mutated. It took over 20 years to identify all these genes, and we will illustrate in this talk how to discover new retinal genes. In IRDs, the retinal-photoreceptor neurons die, the inner retinal layers remain intact for a long time. Rods die first due to the mutation in a crucial retinal gene, followed by apoptosis which leads to nightblindness. Cone death occurs later, through a different mechanism, likely not due to the mutation, but due to oxidative stress, which leads to progressive tunnel vision followed by visual acuity and color loss and complete blindness. The most common form of IRDs are retinitis pigmentosa (RP) in adults and Leber Congenital amaurosis (LCA) in childhood.
Previously, IRDs were deemed untreatable, based on the non-detectable ERGs, the severe loss of acuity, the loss of visual fields and the retinal thinning on OCT. Remarkably, recent human research has shown that this is not true and IRDs respond to a wide variety of treatments, ranging from cell, drug, gene augmentation, gene editing (ASO and CRISPR), to optogenetic interventions. These novel therapies are in various stages of human therapeutic development, ranging from pre-clinical studies to phase I to phase III human trials to FDA approval (e.g. Luxturna, RPE65 gene therapy, 2019). It is important to realize how three major, converging advances in Genetics and Genomics, novel retinal and brain functional testing and imaging, plus animal model therapies have revolutionized this field. Also, how there are multiple treatment avenues in study and in development, increasing the chance of commercial successes and the possibilities of combination treatments.
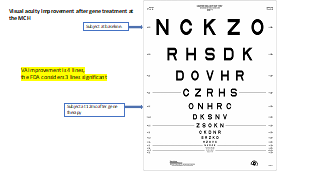
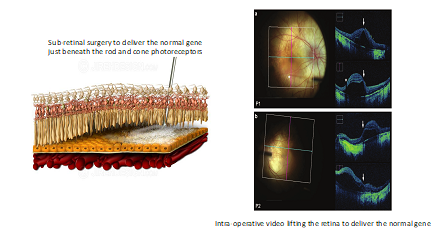
Many research questions remain for the new generation of vision scientists, including which patient and which genotype gets what treatment? When is the best time for intervention? and in combination or alone? For example, gene augmentation is a sub-retinal surgical therapy and is confined to local areas of living cells and is of course gene specific but mutation independent. Gene editing is given intra-vitreally and reaches all retinal cells but is mutation dependent. Atrophic regions need to be treated by stem cells, and/or optogenetic treatments which may be possible in remaining inner retinal cells, when rods and cones have died, while drugs may be efficacious and gene agnostic. Strikingly, it is currently possible to replace at least 5 retinal genes in human studies (i.e. RPE65 in juvenile RP and LCA, CHM in Choroideremia, RPGR in x-linked RP and CNGA3 and CNGB3 in complete achromatopsia, while others are coming). Gene editing is now studied by ASO and CRISPR in CEP290-related LCA and USH2a– related RP and Usher patients (with exon 13 mutations). Optogenetic, stem cell and several drug trials are gene agnostic and have started in humans.
My talk will focus three related endeavors: 1. How to find retinal genes that cause IRDs, my example will be the “candidate gene approach” and how we discovered NMNAT1. 2. Next will be a discussion about how to perform a clinical trial, with the help of novel endpoints, genotyped patients, a whole team of experts and 3. The new registries and data bases containing genotype and phenotype data.
***